计算任务提交(进阶版)
概述
用户在使用SLURM进行计算任务的提交时,可以使用三种不同的方式进行任务提交。
- salloc: 交互式方式,通过提前获取一个资源分配,并进行提交任务
- sbatch: 非交互式方式,直接使用批处理脚本一次提交
- srun: 获取分配的计算资源,并直接运行应用
程序参数
这三种方式可用的参数如下表所示:
- 加粗部分参数为使用频率较高的参数
- 部分参数限定在特定的使用方式中
| 参数 | 说明 | 参数示例 |
|---|---|---|
| --array= | 指定任务列表(只在sbatch 使用) | --array=1-10 |
| --account= | 记账使用的账户 | --account=sonmi |
| --begin= | 在指定时间之后初始任务 | --begin=18:00:00 |
| --cluster= | 指定要提交到的集群(只在sbatch使用) | --cluster=sonmi |
| --constraint= | 要求节点类型,节点的类型在slurm配置文件中设置 | --constraint=haswell |
| --cpus-per-task= | 每个任务请求的CPU数 | --cpus-per-task=4 |
| --dependency=state:jobid | 推迟任务直到jobid的任务转变为state状态 | --dependency=COMPLETED:100 |
| --error= | 错误信息重定向到该文件 | --error=error.log |
| --exclude= | 将指定hostname的节点排除出可分配节点 | --exclude=compute-0-0 |
| --exclusive=[=user] | 分配的节点不与指定的用户共享 | --exclusive=sonmi |
| --export= | 导出指定的环境变量 | --export=MY_ENV=my_env |
| --gres=name[:count] | 每个节点请求的通用资源 | --gres=gpu:1 |
| --input= | 指定从特定文件读取数据 | --input=data.in |
| --label | 在输出文件前添加任务ID(只在srun中使用) | --label=tag |
| --licenses= | 任务请求对应的证书资源 | --licenses=vasp:1 |
| --job-name= | 指定任务的名称 | --job-name=lammps |
| --mem= | 每个节点请求的内存,单位MB | --mem=1000 |
| --mem-per-cpu= | 请求每个CPU各分配对应数量的内存,单位MB | --mem-per-cpu=1000 |
| -N | 任务请求的节点数量 | -N4 |
| -n | 启用的任务数量 | -n2 |
| --nodelist= | 指定要分配任务的节点列表 | --nodelist=compute-0-[0-1],sonmi |
| --ntasks-per-node= | 指定每个节点要分配的线程数 | --ntasks-per-node=4 |
| --output= | 将标准输出重新向到该文件 | --output=output.log |
| --partition= | 指定要分配到的分区或队列 | --partition=sonmi |
| --qos= | 指定服务质量 | --qos=low |
| --signal=B:sig_num:sig_time | 任务运行超时时发送指定信号量给该任务 | --signal=B:SIGUSR1@120(任务超时前120秒发送SIGUSR1信号量) |
| --time= | 任务运行时间限制 | --time=1:00:00 |
| --wrap= | 将指定命令包裹为简单的shell命令(只在sbatch中使用) | --wrap=ls |
后续将使用LAMMPS作为示例应用分别详细讲解这三种使用方式的区别。
示例环境准备
下面的几个例子将以一个简单的LAMMPS分子动力学计算为示例。
- 创建一个项目文件夹:
mkdir ~/lammps-test && cd ~/lammps-testmkdir ~/lammps-test && cd ~/lammps-test- 分别为三种方式创建子文件夹:
mkdir salloc sbatch srunmkdir salloc sbatch srun- 在每个子文件夹中分别创建一个简单的输入文件:
subdir=("salloc" "sbatch" "srun")
for i in ${subdir[@]}; do
cat > $i/lammps.input <<EOF
# 3d Lennard-Jones melt
units lj
atom_style atomic
lattice fcc 0.8442
region box block 0 20 0 20 0 20
create_box 1 box
create_atoms 1 box
mass 1 1.0
velocity all create 1.44 87287 loop geom
pair_style lj/cut 2.5
pair_coeff 1 1 1.0 1.0 2.5
neighbor 0.3 bin
neigh_modify delay 5 every 1
fix 1 all nve
run 200000
EOF
donesubdir=("salloc" "sbatch" "srun")
for i in ${subdir[@]}; do
cat > $i/lammps.input <<EOF
# 3d Lennard-Jones melt
units lj
atom_style atomic
lattice fcc 0.8442
region box block 0 20 0 20 0 20
create_box 1 box
create_atoms 1 box
mass 1 1.0
velocity all create 1.44 87287 loop geom
pair_style lj/cut 2.5
pair_coeff 1 1 1.0 1.0 2.5
neighbor 0.3 bin
neigh_modify delay 5 every 1
fix 1 all nve
run 200000
EOF
done- tree命令查看项目目录情况:
[sonmi@sonmi lammps-test]$ tree
.
├── salloc
│ └── lammps.input
├── sbatch
│ └── lammps.input
└── srun
└── lammps.input[sonmi@sonmi lammps-test]$ tree
.
├── salloc
│ └── lammps.input
├── sbatch
│ └── lammps.input
└── srun
└── lammps.inputsalloc:交互式提交任务
salloc的使用基本步骤为申请计算节点,然后提交任务到对应的计算节点。下面分别对单节点和跨节点这两种方式进行说明。
单节点运行
进入示例文件夹:
cd ~/lammps-test/salloccd ~/lammps-test/salloc先申请一个可分配4个核心的节点:
salloc -p sonmi -N1 -n4 -q low -t 1:00:00salloc -p sonmi -N1 -n4 -q low -t 1:00:00该命令中:
-p: 指定SonmiHPC的默认sonmi分区;
-N1: 指定了请求一个计算节点;
-n4: 参数请求4个CPU核心资源;
-q low: 参数设置qos为low,既较低的服务质量;
-t 1:00:00: 限制了任务的最长运行时间为1小时。
请求的返回结果如下所示,可以看到已经分配了compute-0-0节点,并且任务id为3:
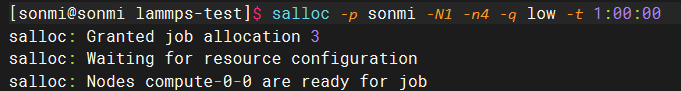
然后通过ssh到对应节点执行命令即可:
ssh compute-0-0 # 进入到分配的对应计算节点
cd ~/lammps-test/salloc # 进入项目文件夹
source /opt/intel/oneapi/setvars.sh # 激活并行计算环境变量
mpirun -np 4 /opt/sonmi/bin/lmp -in lammps.input # 命令行运行计算程序ssh compute-0-0 # 进入到分配的对应计算节点
cd ~/lammps-test/salloc # 进入项目文件夹
source /opt/intel/oneapi/setvars.sh # 激活并行计算环境变量
mpirun -np 4 /opt/sonmi/bin/lmp -in lammps.input # 命令行运行计算程序运行结束后,使用如下命令取消任务即可,任务ID为上面请求得到的任务ID:
scancel 3scancel 3跨节点运行
先申请两个计算节点,每个节点4个进程:
salloc -p sonmi -N2 --ntasks-per-node=4 -q low -t 1:00:00salloc -p sonmi -N2 --ntasks-per-node=4 -q low -t 1:00:00其中--ntasks-per-node可以用于指定在分配的两个节点中如何分配进程,示例中每个节点执行4个进程,一个进程相当于一个CPU核心数。返回的结果如下面所示:
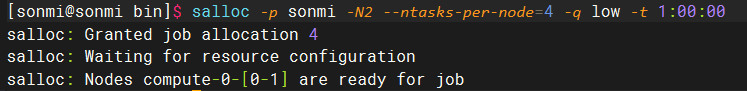
在项目目录中执行如下命令生成hostfile文件:
srun hostname -s| sort -n > hostfilesrun hostname -s| sort -n > hostfile使用如下的命令执行并行程序:
mpirun -np 8 -machinefile hostfile /opt/sonmi/bin/lmp -in lammps.inputmpirun -np 8 -machinefile hostfile /opt/sonmi/bin/lmp -in lammps.inputsbatch:批处理脚本提交任务
使用sbatch批处理脚本来提交任务是用得相对较多并且推荐的首要方式。sbatch脚本基于linux shell脚本,通过在脚本前添加特定的注释字段来让SLURM调度器识别特定参数,具体的脚本内容与Shell脚本一致,因此可以在其中添加自己的额外程序逻辑。
sbatch参数使用#SBATCH <param>的形式在脚本开头提供。可以使用的参数如上面程序参数部分。
在sbatch提交脚本中,可以使用以下的内置环境变量:
| 环境变量 | 说明 |
|---|---|
| SLURM_ARRAY_JOB_ID | 如果该任务是任务列表中的一个,该变量为对应的job ID |
| SLURM_ARRAY_TASK_ID | 如果该任务是任务列表中的一个,该变量为对应task ID |
| SLURM_CLUSTER_NAME | 该任务所分配到的集群名称 |
| SLURM_CPUS_PER_TASK | 该任务每个task所请求的CPU核心数 |
| SLURM_JOB_ACCOUNT | 该任务的账户名称 |
| SLURM_JOB_ID | 该任务的ID |
| SLURM_JOB_NAME | 该任务的名称 |
| SLURM_JOB_NODELIST | 分配给该任务的节点列表 |
| SLURM_JOB_NUM_NODES | 分配给该任务的节点数量 |
| SLURM_JOB_PARTITION | 运行该任务的分区或队列 |
| SLURM_JOB_UID | 任务所有者的UID |
| SLURM_JOB_USER | 任务所有者的用户名 |
| SLURM_RESTART_COUNT | 任务重启次数 |
| SLURM_PROCID | 进程对应的MPI的秩 |
| SLURM_STEP_ID | 任务步ID |
| SLURM_STEP_NUM_TASKS | task数量,既MPI程序的秩数量 |
| SLURM_SUBMIT_DIR | 任务提交目录 |
| SLURM_CPUS_ON_NODE | 节点所拥有的CPU核心数 |
以上的环境变量均可以在sbatch脚本中使用。
下面将使用sbatch方式提交一个计算任务,该计算任务请求2个计算节点,每个节点请求4个CPU核心,并在计算开始前打印上面其中的一些环境变量。
先进入项目目录中:
cd ~/lammps-test/sbatchcd ~/lammps-test/sbatch使用如下命令,创建该任务的sbatch脚本:
cat > lmp.sub <<EOF
#!/bin/bash
#SBATCH --job-name=lammps
#SBATCH --partition=sonmi
#SBATCH --nodelist=compute-0-[0-1]
#SBATCH --ntasks-per-node=4
#SBATCH --output=%j.out
#SBATCH --error=%j.err
#SBATCH --exclusive
#SBATCH --time=1:00:00
ulimit -s unlimited
ulimit -l unlimited
source /opt/intel/oneapi/setvars.sh
cd $SLURM_SUBMIT_DIR
echo "SLURM_JOB_NAME: \${SLURM_JOB_NAME}"
echo "SLURM_JOB_NODELIST: \${SLURM_JOB_NODELIST}"
echo "SLURM_JOB_ID: \${SLURM_JOB_ID}"
mpirun -np 8 /opt/sonmi/bin/lmp -in \$SLURM_SUBMIT_DIR/lammps.input
EOFcat > lmp.sub <<EOF
#!/bin/bash
#SBATCH --job-name=lammps
#SBATCH --partition=sonmi
#SBATCH --nodelist=compute-0-[0-1]
#SBATCH --ntasks-per-node=4
#SBATCH --output=%j.out
#SBATCH --error=%j.err
#SBATCH --exclusive
#SBATCH --time=1:00:00
ulimit -s unlimited
ulimit -l unlimited
source /opt/intel/oneapi/setvars.sh
cd $SLURM_SUBMIT_DIR
echo "SLURM_JOB_NAME: \${SLURM_JOB_NAME}"
echo "SLURM_JOB_NODELIST: \${SLURM_JOB_NODELIST}"
echo "SLURM_JOB_ID: \${SLURM_JOB_ID}"
mpirun -np 8 /opt/sonmi/bin/lmp -in \$SLURM_SUBMIT_DIR/lammps.input
EOF之后执行脚本即可:
sbatch lmp.subsbatch lmp.sub提交后任务执行后可以看到如下的输出,对应的环境变量也打印了出来:
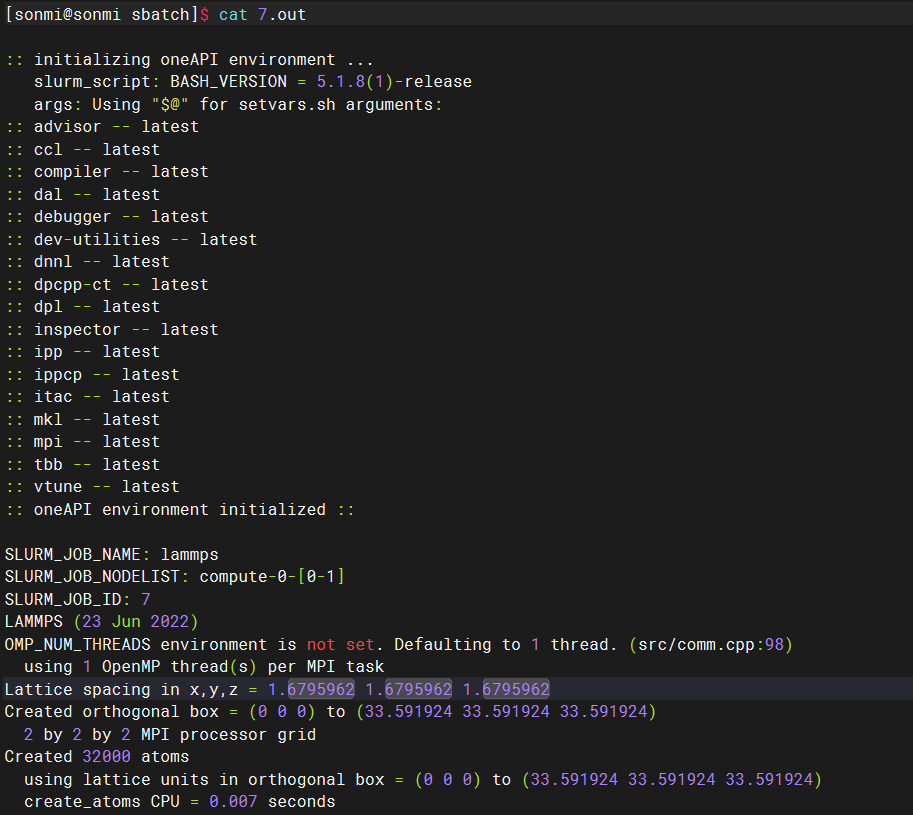
srun: 即时分配资源并提交任务
有时候对一些简单的任务,不想写sbatch提交脚本,可以通过srun来提交运行。
首先先进入项目目录中:
cd ~/lammps-test/sruncd ~/lammps-test/srun激活MPI并行环境:
source /opt/intel/oneapi/setvars.shsource /opt/intel/oneapi/setvars.sh使用srun命令提交计算任务:
srun --nodes=2 --ntasks-per-node=4 mpirun -np 8 /opt/sonmi/bin/lmp -in lammps.inputsrun --nodes=2 --ntasks-per-node=4 mpirun -np 8 /opt/sonmi/bin/lmp -in lammps.input该命令中请求2个计算节点,每个节点请求4个task。