
产品介绍 ST-QY36 是一款4U 双路36盘位存储服务器,支持多种传输协议,支持SSD缓存,是搭…

服务热线
王经理 17727752803
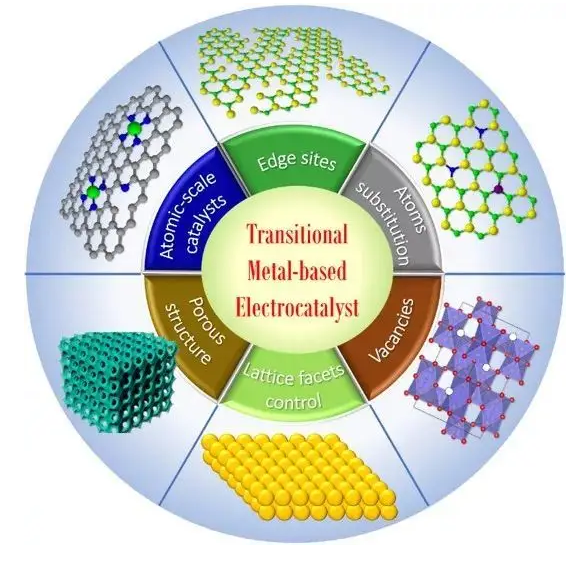
公司依托于强大的高校科研技术体系及世界前沿高科技应用技术,专注于高性能计算技术(High Performance Computing)和数据安全存储技术应用级整体解决方案。公司坚持“顾问式一站服务”,是高性能“并行”计算集群&安全存储应用级整体解决方案服务商,为科研工作者和AI创业人员提供超算算力(包括算力租赁)、通用算力、定制化算力、异构算力、数据安全存储以及相关配套服务的IT软、硬件整体解决、实施方案。
专业的高性能计算软硬件定制为客户提供超算算力整套专业化方案,数据安全存储及算力软、硬件使用专业化咨询服务。
灵活提供各学科计算解决方案致力于量子化学计算、分子设计、分子模拟、材料设计、生信计算等领域。
专业靠谱的售后服务24*365售后运维体系,顾问式一站服务,一对一责任制及终身技术支持。

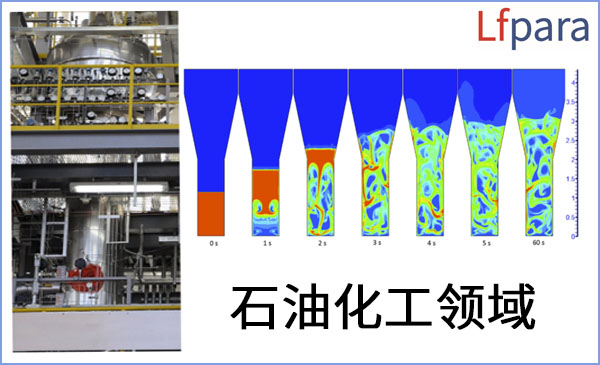
根据客户具体科研需求及预算,为客户提供高性能计算机(服务器)软硬件,HPC集群方案设计及搭建,HPC技术服务承包,海量及高吞吐I/O数据存储,定制化AI算力等。

提供存储、存储网络、存储服务器等解决方案,分步式、嵌入式架构存储产品,混合云存储网络架构整体解决方案及技术实施支持,基于X86、非X86架构横向、纵向灵 活扩展的分布式存储整体解决方案

提供计算化学、计算材料学、计算固体/流体力学、工业仿真等领域的委托计算服务,计算化学、计算材料学、计算力学、HPC维护、HPC工程师培训等及HPC计算机集群硬件及网络知识培训。

资深Win、Linux系统运维工程师团 队,大部分技术提供免费服务,国内外HPC专家技术团队一对一服务,较多高校课题组合作伙伴,集中于计算领域专家数名。

产品介绍 ST-QY36 是一款4U 双路36盘位存储服务器,支持多种传输协议,支持SSD缓存,是搭…

产品介绍 RD-2U60 是一款2U 双路高性能服务器,极强的拓展性,最高支持两张双宽GPU拓展。 …

超静音,支持双路 Scalable处理器,8个热插拔盘位,双GPU,适合无机房条件的课题组。

支持双路 Scalable处理器,8个热插拔盘位,四GPU。适合GPU密集型计算,需机房环境。

四子星,单节点支持双路 Scalable处理器,适合CPU密集型计算,需机房环境。

支持四路 Scalable处理器,双GPU。适合各种计算需求,需机房环境。
一路上,我们更在意如何积累和沉淀
